| SCS【41】基于贝叶斯反卷积法整合分析bulk和scRNA | 您所在的位置:网站首页 › scrna seq和snrnaseq › SCS【41】基于贝叶斯反卷积法整合分析bulk和scRNA |
SCS【41】基于贝叶斯反卷积法整合分析bulk和scRNA
|
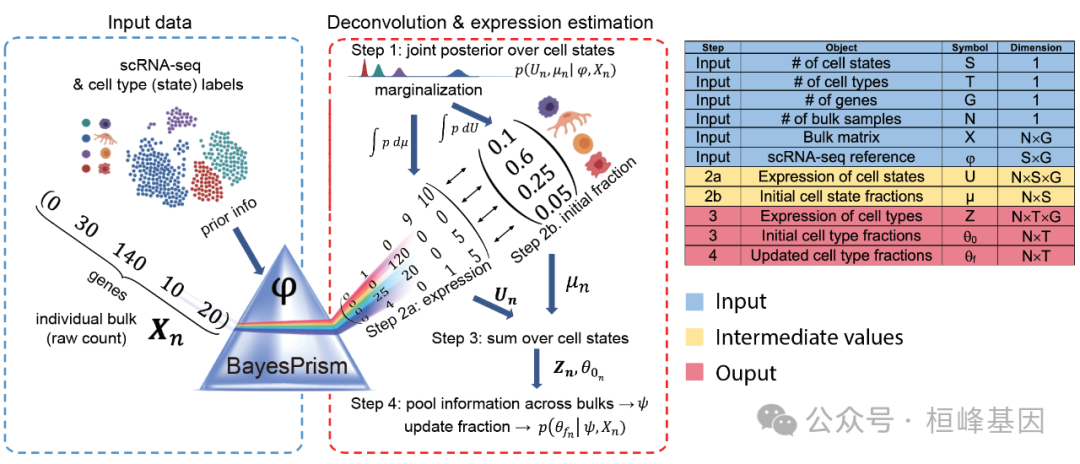
BayesPrism 使用从匹配或相似组织类型收集的scRNA-seq样本,对大量RNA-seq(和空间转录组学)进行细胞类型和基因表达反褶积。将scRNA-seq作为先验信息,估计P(θ,Z|X,ϕ),即细胞类型分数θ和细胞类型特异性基因表达Z在每个群体中的联合后验分布,条件是参考ϕ和每个观察群体X。
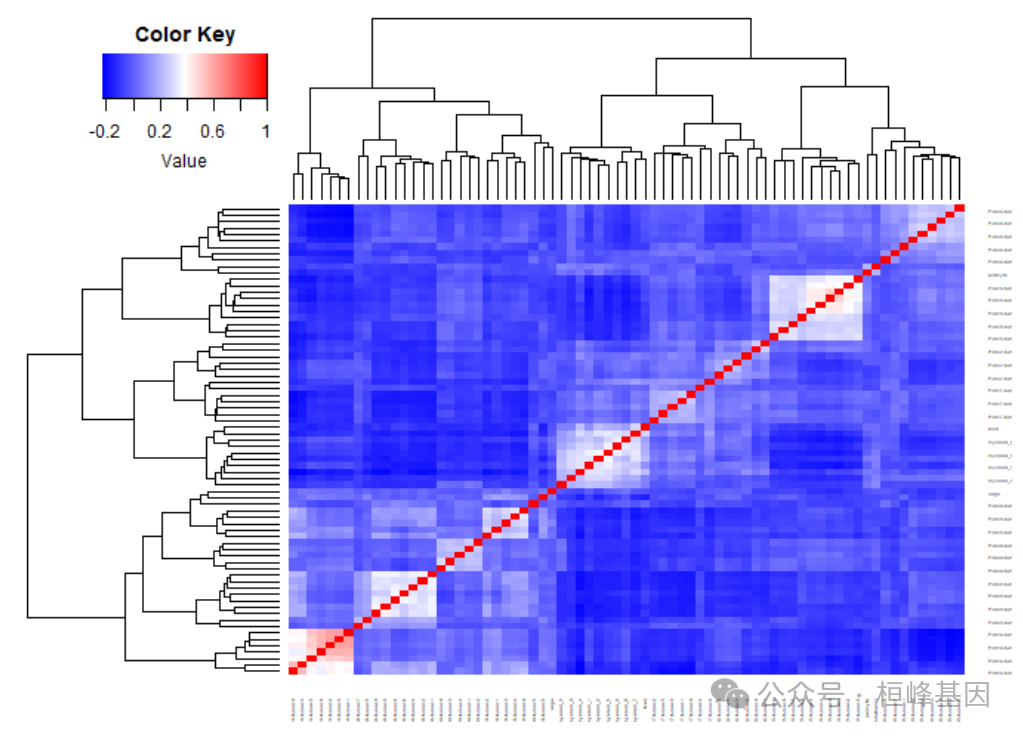
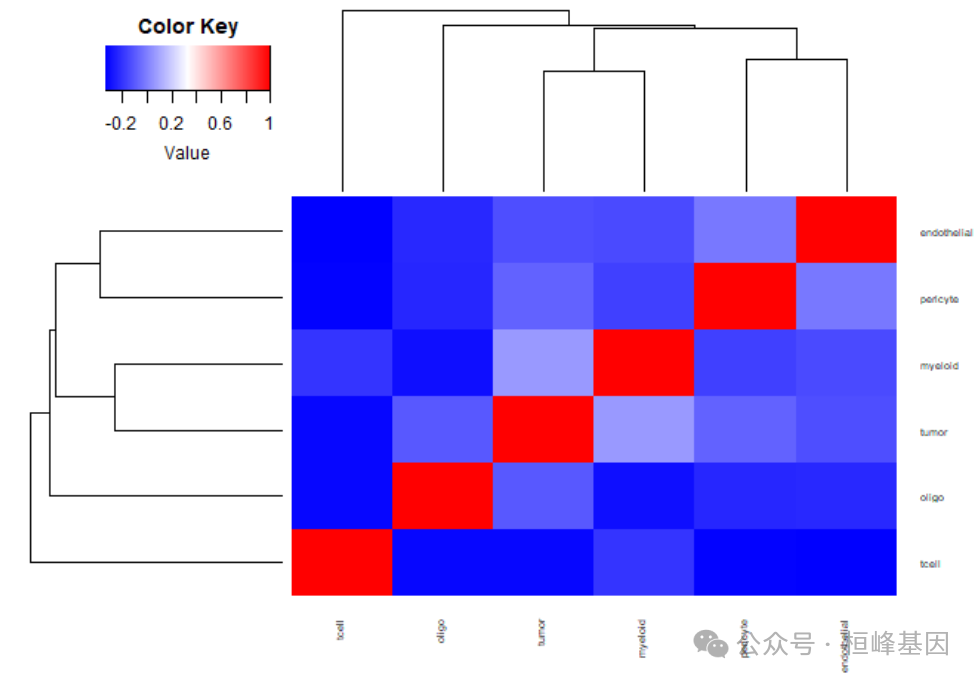
使用BayesPrism对大量RNA-seq数据集TCGA-GBM进行反卷积,使用Yuan等人从8个保留队列中收集的scRNA-seq数据集。 rdata文件包含运行BayesPrism所需的四个对象: bk.dat:批量RNA-seq表达的样本按基因原始计数矩阵。行名是样本id,而列名是基因名。 sc.dat:单细胞RNA-seq表达的细胞-基因原始计数矩阵。行名是细胞id,而列名是基因名。data应该是一个矩阵。如果您的sc.dat是一个稀疏矩阵(dgCMatrix),则应该将其转换为密集矩阵。 cell.type.labels:是一个与nrow(sc.dat)长度相同的字符向量,用于表示引用中每个细胞类型。 cell.state.labels:是一个与nrow(sc.dat)长度相同的字符向量,用于表示引用中每个细胞的细胞状态。在我们的例子中,通过对每个患者的恶性细胞进行亚聚类来获得恶性细胞的细胞状态,通过对所有患者的髓细胞进行聚类来获得髓细胞的细胞状态。为这两种细胞类型定义了多个细胞状态,因为包含大量异质性,同时也有足够数量的细胞用于亚聚类。 library(BayesPrism) source("run_gibbs.R") load("BayesPrism-main/tutorial.dat/tutorial.gbm.rdata") ls() ## [1] "bk.dat" "cell.state.labels" "cell.type.labels" ## [4] "run.gibbs.refPhi" "sc.dat" dim(bk.dat) ## [1] 169 60483 head(rownames(bk.dat)) ## [1] "TCGA-06-2563-01A-01R-1849-01" "TCGA-06-0749-01A-01R-1849-01" ## [3] "TCGA-06-5418-01A-01R-1849-01" "TCGA-06-0211-01B-01R-1849-01" ## [5] "TCGA-19-2625-01A-01R-1850-01" "TCGA-19-4065-02A-11R-2005-01" head(colnames(bk.dat)) ## [1] "ENSG00000000003.13" "ENSG00000000005.5" "ENSG00000000419.11" ## [4] "ENSG00000000457.12" "ENSG00000000460.15" "ENSG00000000938.11" dim(sc.dat) ## [1] 23793 60294 head(rownames(sc.dat)) ## [1] "PJ016.V3" "PJ016.V4" "PJ016.V5" "PJ016.V6" "PJ016.V7" "PJ016.V8" head(colnames(sc.dat)) ## [1] "ENSG00000130876.10" "ENSG00000134438.9" "ENSG00000269696.1" ## [4] "ENSG00000230393.1" "ENSG00000266744.1" "ENSG00000202281.1" sort(table(cell.type.labels)) ## cell.type.labels ## tcell oligo pericyte endothelial myeloid tumor ## 67 160 489 492 2505 20080 sort(table(cell.state.labels)) ## cell.state.labels ## PJ017-tumor-6 PJ032-tumor-5 myeloid_8 PJ032-tumor-4 PJ032-tumor-3 ## 22 41 49 57 62 table(cbind.data.frame(cell.state.labels, cell.type.labels)) ## cell.type.labels ## cell.state.labels endothelial myeloid oligo pericyte tcell tumor ## endothelial 492 0 0 0 0 0 ## myeloid_0 0 550 0 0 0 0 ## myeloid_1 0 526 0 0 0 0 ## myeloid_2 0 386 0 0 0 0 ## oligo 0 0 160 0 0 0 ## pericyte 0 0 0 489 0 0 ## PJ016-tumor-0 0 0 0 0 0 621 ## PJ016-tumor-1 0 0 0 0 0 619 ## tcell 0 0 0 0 67 0 实例操作 细胞类型和状态的质量控制首先绘制细胞状态和细胞类型之间的两两相关矩阵,了解其的质量情况。在细胞类型/状态没有足够数量的信息表示的情况下(低细胞计数和/或低库大小),低质量的细胞类型/状态倾向于聚集在一起。可以以更高的粒度重新聚类数据,或者将这些细胞类型/状态与最相似的细胞类型/状态合并,或者删除。 细胞状态统计 ##QC of cell type and state labels plot.cor.phi (input=sc.dat, input.labels=cell.state.labels, title="cell state correlation", #specify pdf.prefix if need to output to pdf #pdf.prefix="gbm.cor.cs", cexRow=0.2, cexCol=0.2, margins=c(2,2))
核糖体蛋白基因和线粒体基因等表达量较大的基因可能在分布中占主导地位,并对推断产生偏差。这些基因通常不能提供区分细胞类型的信息,而且可能是大量虚假变异的来源。因此,不利于反褶积,建议去除这些基因。 可视化从scRNA-seq参考的异常基因的分布。计算每个基因在所有细胞类型中的平均表达量和细胞类型特异性评分。 可视化和确定scRNA-seq数据中的异常基因 如图所示,核糖体蛋白基因通常表现出高平均表达和低细胞类型特异性评分。 sc.stat |

【本文地址】